
Blood丨闵军霞/王福俤团队发现铁泵蛋白FPN降解调控关键机制
近日,国际权威学术期刊《血液学》(Blood)(影响因子:17.543)在线发表了浙江大学转化医学研究院闵军霞教授/浙江大学医学院王福俤教授团队题为“RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation”的研究论文[1],首次阐明了铁泵蛋白(Ferroportin,FPN)在体内外被降解的调控机制。
论文链接:
该研究筛选并发现了新型E3泛素连接酶RNF217介导了FPN的降解,于国际上率先制备了条件性敲除RNF217的小鼠模型,并首次揭示了RNF217通过调控FPN降解进而维持机体铁稳态代谢的新机制。此外,该研究通过转录组及甲基化测序的方法首次揭秘了DNA去甲基化明星分子Tet1响应机体铁紊乱,并通过表观遗传学调控RNF217-FPN表达,进而控制机体对于高铁环境的响应。
这一重磅发现不仅攻克了铁代谢领域多年来尚未被解析的难点问题,同时为铁紊乱相关疾病的防治提供了新靶点和新思路。

图1:论文首页截图
铁元素不仅是血红蛋白、肌红蛋白合成的主要原料,也是多种氧化还原酶类的辅因子。作为人体含量最多的必需微量元素,铁稳态代谢对维持生命健康至关重要。原发性遗传性血色病是铁过载人类遗传病,主要由HFE、HJV、TfR2、HAMP或FPN等铁代谢调控基因突变引发多器官铁蓄积,多累及损伤心脏、肝脏和胰腺等重要脏器,患者晚期多并发心脏病、肝硬化或糖尿病等疾病,严重危害机体健康[2]。
FPN是迄今为止唯一已知的细胞铁离子外排蛋白,它在十二指肠上皮细胞、小肠树突状细胞、巨噬细胞以及肝实质细胞中高表达,其中小肠上皮细胞和巨噬细胞表达最为丰富;FPN通过控制铁离子的吸收、再循环和储存来维持机体铁稳态。课题组前期利用基因敲除小鼠在国际上首先发现并报道了FPN在巨噬细胞以及肝实质细胞中调控铁离子储存和再循环的重要作用,并揭示FPN是上述组织细胞中外排铁离子的关键通道蛋白[3,4,5]。为了维持铁离子代谢平衡从而保障细胞和组织功能,FPN的表达受到精密调控。铁调素Hepcidin(HAMP基因编码蛋白)是肝脏分泌的调控铁稳态的核心激素,其作用靶点是FPN。当Hepcidin与FPN结合时,FPN降解而失去外排铁离子功能,进而抑制十二指肠上皮细胞的铁吸收及巨噬细胞的铁再循环。一旦HAMP表达被抑制或FPN功能失调,导致铁外排功能异常,将引发机体铁代谢紊乱疾病[6]。因此,Hepcidin结合FPN并诱导其降解,是FPN蛋白维持铁稳态代谢的根本机制[7]。早期研究提出,泛素化在FPN的降解过程中发挥关键作用[8]。
然而,结合并降解FPN的E3泛素化连接酶至今是困扰领域的重大难题。王福俤和闵军霞团队通过多年努力,发现并锁定RNF217是调控FPN泛素化降解的重要蛋白。
表观遗传修饰在调节染色质结构、调控基因表达、维持细胞稳态等生命进程中扮演重要角色。课题组前期研究表明,表观遗传调控同样参与了机体铁稳态的维持[9,10]。为了明确机体铁代谢紊乱过程中的关键表观遗传调控分子,该研究通过对高低铁膳食饲喂的铁紊乱小鼠模型进行全基因组甲基化测序及转录组测序筛选,发现DNA去甲基化酶Tet1参与了小鼠对于高铁环境的响应。
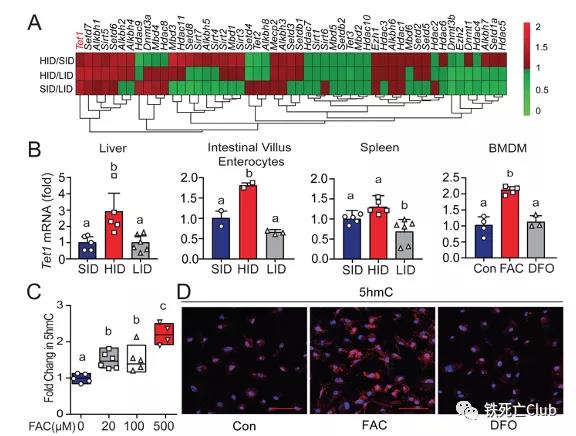
图2:Tet1参与小鼠对于高铁刺激的响应
(选自论文,Figure 1)
进一步给予Tet1敲除小鼠高铁刺激,该小鼠出现类似血色病的表型,主要表现为血清铁及转铁蛋白饱和度升高,肝脏铁增加,然而脾脏及小肠上皮细胞铁离子减少的现象。通过检测铁调素Hepcidin及其他铁代谢调节基因表达水平,研究者发现Tet1缺失导致FPN蛋白在小肠上皮细胞及巨噬细胞蓄积,进而引发小鼠铁代谢紊乱。有趣的是,Tet1并非直接通过调控Hepcidin或FPN的转录来介导机体的铁稳态代谢。
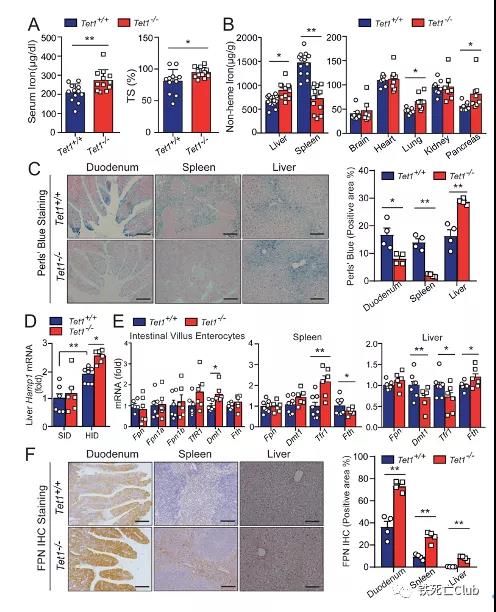
图3:Tet1基因敲除小鼠铁代谢紊乱
(选自论文,Figure 2)
为了深入探究Tet1调控FPN表达的分子机制,研究者检测小鼠原代巨噬细胞中FPN降解情况,发现Tet1缺乏导致FPN蛋白蓄积、泛素化水平减弱,表明Tet1通过泛素化降解路径调控FPN表达。然而,FPN如何被降解仍属于铁代谢领域的未知。为明确既能促进FPN泛素化降解又能被Tet1调控的关键蛋白,该研究采用双向筛选的方法:利用酵母双杂交技术在E3连接酶文库中筛选与FPN结合的蛋白,并进一步在哺乳动物细胞中验证相互作用;同时在Tet1敲除体系的数据库中筛选潜在的靶基因。通过基因叠加分析及后续的表观遗传修饰分析,研究者筛选出既能被Tet1调控,又能与FPN结合的E3连接酶RNF217。
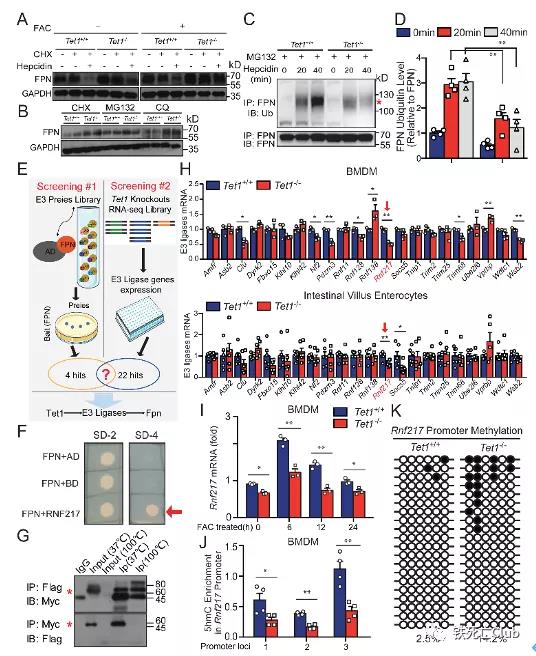
图4:RNF217可能是Tet1调控的降解FPN的E3连接酶
(选自论文,Figure 4)
那么RNF217是否调控了FPN的泛素化降解呢?为了回答这一问题,研究者首先在体外检测了RNF217对于FPN的调控作用,结果显示外源RNF217显著促进了蛋白酶体及溶酶体依赖的FPN的泛素化降解,功能性缺失或突变RNF217的酶活位点,该降解作用明显被抑制。上述结果揭示了FPN是RNF217的作用靶点。
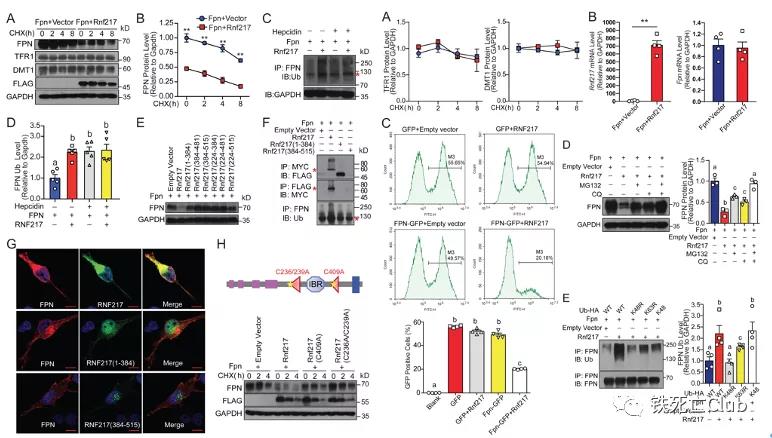
图5:RNF217促进FPN降解
(选自论文,Figure 5,Figure S5)
为了进一步明确RNF217的生理功能,研究者分别构建了巨噬细胞和小肠上皮细胞特异性缺失RNF217的基因敲除小鼠(Rnf217Lysm/Lysm和Rnf217Villin/Villin)。令人惊奇的是,Rnf217Lysm/Lysm和Rnf217Villin/Villin小鼠表现出与Tet1敲除小鼠类似的对高铁响应异常的铁代谢紊乱表型,表明其在铁稳态代谢调控中的重要作用。通过分离小鼠原代巨噬细胞,研究者发现RNF217缺失导致FPN降解减弱。由于Hepcidin是诱发FPN发生内吞并降解的重要分子,为揭示RNF217在Hepcidin-FPN调控轴中的关键作用,研究者给予Rnf217Lysm/Lysm小鼠注射Hepcidin或Hepcidin激活剂LPS,结果显示RNF217缺失显著抑制了Hepcidin依赖的FPN降解。上述结果既明确了体内RNF217在Hepcidin调控的FPN降解中的重要作用,同时提示RNF217有望成为Hepcidin-FPN相关铁代谢紊乱疾病的治疗靶点。
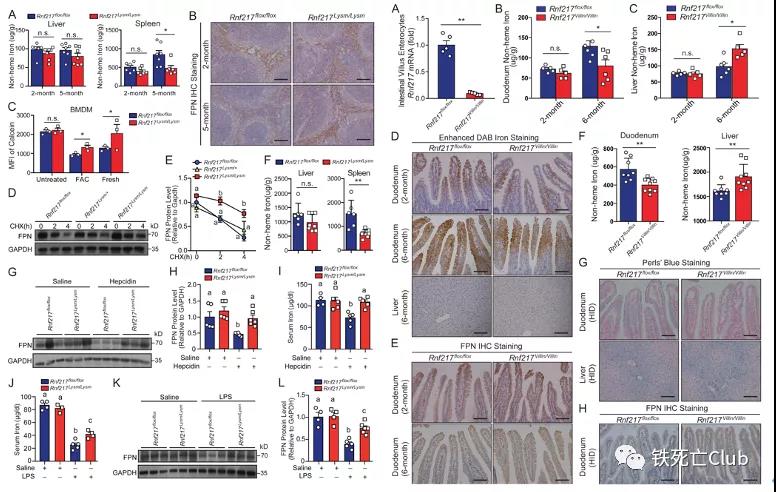
图6:巨噬细胞和小肠上皮细胞特异性敲除RNF217
(选自论文,Figure 6、7)
总之,这项研究于国际上首次发现并报道了调控FPN降解的E3泛素连接酶RNF217。该成果成功制备了Rnf217条件性敲除小鼠,于体内外探索并揭示了Rnf217调控FPN降解及铁稳态的分子机制;该成果提出了Tet1-Rnf217-FPN轴响应机体高铁负荷的新机制,不仅填补了铁代谢研究领域关于FPN降解调控的空白,同时为铁代谢紊乱相关疾病的防治提供了新靶点和新策略。
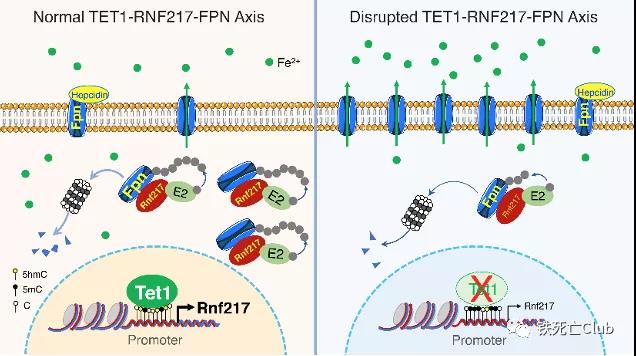
图7:Tet1-Rnf217-FPN调控轴模式图
王福俤团队博士后蒋丽和博士生王佳明为论文共同第一作者;浙江大学王福俤教授、闵军霞教授为共同通讯作者。
此项研究得到中国科学院生物化学与细胞生物学研究所胡荣贵研究员、浙江大学生命科学研究院金建平研究员、浙江大学医学院孙启明教授、浙江大学实验动物中心洪胜辉以及医学院公共技术平台陈静瑶的大力帮助。项目受到国家重点研发计划、国家自然科学基金以及中国博士后科学基金的经费资助。

图8:论文主要作者
闵军霞教授、王福俤教授团队合影
参考文献
1.Jiang L, Wang J, Wang K, et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood. 2021 Apr 25;blood.2020008986. doi: 10.1182/blood.2020008986. Online ahead of print.
2.Prabhu A, Cargill T, Roberts N, Ryan JD. Systematic Review of the Clinical Outcomes of Iron Reduction in Hereditary Hemochromatosis. Hepatology (Baltimore, Md). 2020;72(4):1469-1482.
3.Zhang Z, Zhang F, An P, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood. 2011;118(7):1912-1922.
4.Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology (Baltimore, Md). 2012;56(3):961-971.
5.Bessman NJ, Mathieu JRR, Renassia C, et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science. 2020;368(6487):186-189.
6.Drakesmith H, Nemeth E, Ganz T. Ironing out Ferroportin. Cell metabolism. 2015;22(5):777-787.
7.Billesbølle CB, Azumaya CM, Kretsch RC, et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature. 2020;586(7831):807-811.
8.Qiao B, Sugianto P, Fung E, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell metabolism. 2012;15(6):918-924.
9.Yin X, Wu Q, Monga J, et al. HDAC1 Governs Iron Homeostasis Independent of Histone Deacetylation in Iron-Overload Murine Models. Antioxidants & redox signaling. 2018;28(13):1224-1237.
10.Tao Y, Wu Q, Guo X, Zhang Z, Shen Y, Wang F. MBD5 regulates iron metabolism via methylation-independent genomic targeting of Fth1 through KAT2A in mice. British journal of haematology. 2014;166(2):279-291.
